Setup the Seurat Object
For this tutorial, we will be analyzing the a dataset of Peripheral Blood Mononuclear Cells (PBMC) freely available from 10X Genomics. There are 2,700 single cells that were sequenced on the Illumina NextSeq 500. The raw data can be found here.
library(dplyr)
library(Seurat)
library(patchwork)
library(gridExtra)
# Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "Data/hg19/")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc
Attaching package: 'dplyr'
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
Loading required package: SeuratObject
Loading required package: sp
'SeuratObject' was built under R 4.3.1 but the current version is
4.3.2; it is recomended that you reinstall 'SeuratObject' as the ABI
for R may have changed
'SeuratObject' was built with package 'Matrix' 1.6.3 but the current
version is 1.6.5; it is recomended that you reinstall 'SeuratObject' as
the ABI for 'Matrix' may have changed
Attaching package: 'SeuratObject'
The following object is masked from 'package:base':
intersect
Attaching package: 'gridExtra'
The following object is masked from 'package:dplyr':
combine
Warning message:
"Feature names cannot have underscores ('_'), replacing with dashes ('-')"
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
1 layer present: counts
What does data in a count matrix look like?
# Lets examine a few genes in the first thirty cells
pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]
[[ suppressing 30 column names 'AAACATACAACCAC-1', 'AAACATTGAGCTAC-1', 'AAACATTGATCAGC-1' ... ]]
3 x 30 sparse Matrix of class "dgCMatrix"
CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .
dense.size <- object.size(as.matrix(pbmc.data))
dense.size
709591472 bytes
sparse.size <- object.size(pbmc.data)
sparse.size
29905192 bytes
dense.size/sparse.size
23.7 bytes
Standard pre-processing workflow
Selection and filtration base on QC-metrics, data normalization and scaling, and the dectection of highly variable features
QC and setecting cells for further data analysis
Quality Control Metrics
- The number of unique genes detected in each cell.
- Low-quality cells or empty droplets will often have very few genes
- Cell doublets or multiplets may exhibit an aberrantly high gene count
- Similarly, the total number of molecules detected within a cell (correlates strongly with unique genes)
- The percentage of reads that map to the mitochondrial genome
- Low-quality / dying cells often exhibit extensive mitochondrial contamination
- We calculate mitochondrial QC metrics with the PercentageFeatureSet() function, which calculates the percentage of counts originating from a set of features
- We use the set of all genes starting with MT- as a set of mitochondrial genes
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
Where are QC metrics stored in Seurat?
- The number of unique genes and total molecules are automatically calculated during
CreateSeuratObject()- You can find them stored in the object meta data
# Show QC metrics for the first 5 cells
head(pbmc@meta.data, 5)
| orig.ident | nCount_RNA | nFeature_RNA | percent.mt | |
|---|---|---|---|---|
| <fct> | <dbl> | <int> | <dbl> | |
| AAACATACAACCAC-1 | pbmc3k | 2419 | 779 | 3.0177759 |
| AAACATTGAGCTAC-1 | pbmc3k | 4903 | 1352 | 3.7935958 |
| AAACATTGATCAGC-1 | pbmc3k | 3147 | 1129 | 0.8897363 |
| AAACCGTGCTTCCG-1 | pbmc3k | 2639 | 960 | 1.7430845 |
| AAACCGTGTATGCG-1 | pbmc3k | 980 | 521 | 1.2244898 |
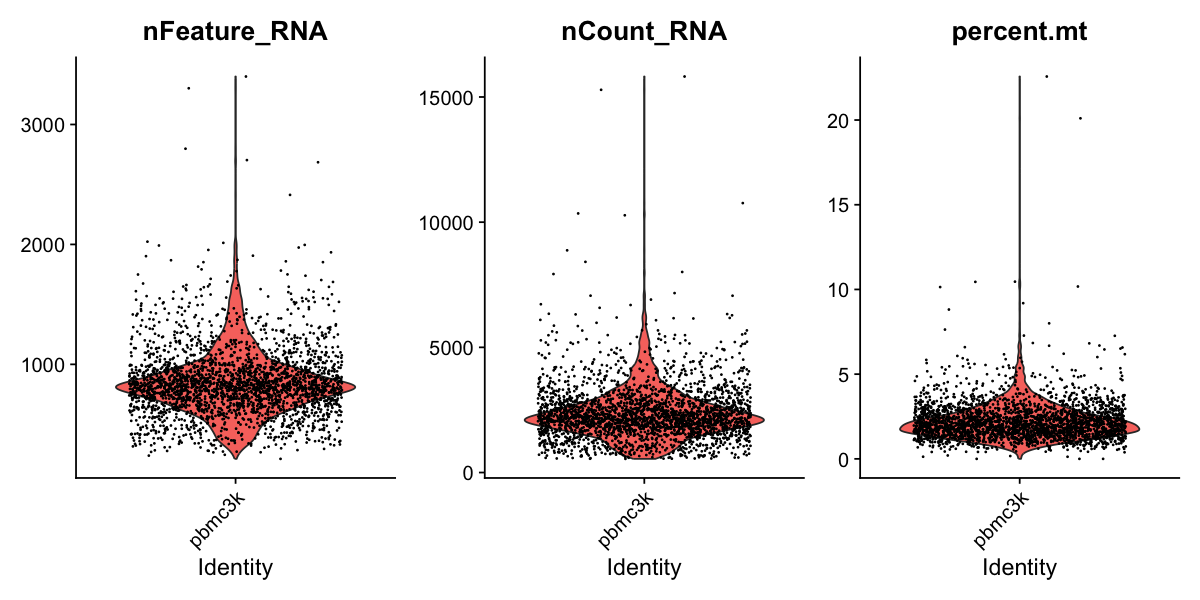
In the example below, we visualize QC metrics, and use these to filter cells.
- We filter cells that have unique feature counts over 2,500 or less than 200
- We filter cells that have >5% mitochondrial counts
# Visualize QC metrics as a violin plot
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
options(repr.plot.width = 10, repr.plot.height = 5)
Warning message:
"Default search for "data" layer in "RNA" assay yielded no results; utilizing "counts" layer instead."
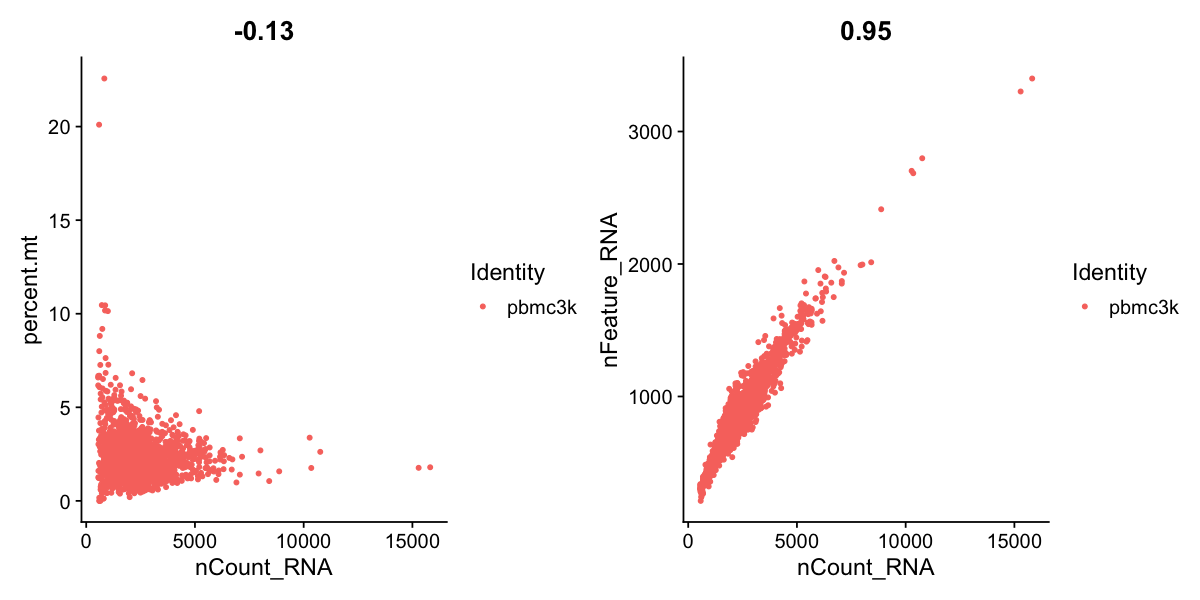
# FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
Normalizing the data
After removing unwanted cells from the dataset, the next step is to normalize the data. By default, we employ a global-scaling normalization method “LogNormalize” that normalizes the feature expression measurements for each cell by the total expression, multiplies this by a scale factor (10,000 by default), and log-transforms the result. In Seurat v5, Normalized values are stored in pbmc[["RNA"]]$data.
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
# the previous command show the default parameters, it is equivalent with
# pbmc <- NormalizeData(pbmc)
Normalizing layer: counts
SCTransform()
for more information check out SCTransform() or here.
The use of SCTransform replaces the need to run NormalizeData, FindVariableFeatures, or ScaleData (described below.)
Feature Selection: Identification of highly variable features
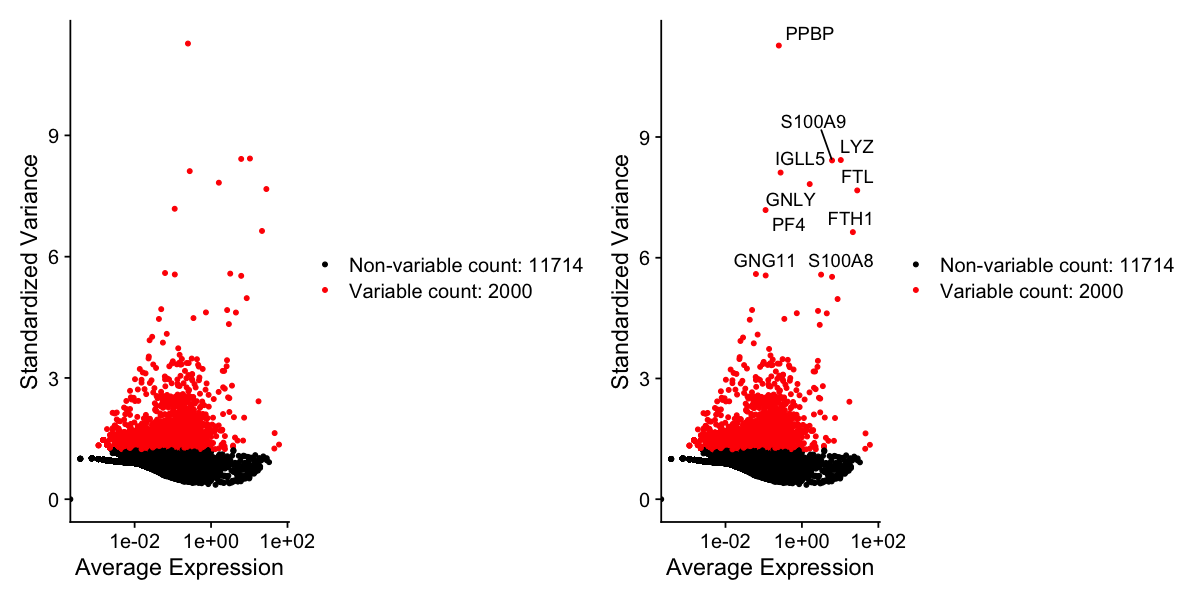
We next calculate a subset of features that exhibit high cell-to-cell variation in the dataset (i.e, they are highly expressed in some cells, and lowly expressed in others). We and others have found that focusing on these genes in downstream analysis helps to highlight biological signal in single-cell datasets.
FindVariableFeatures() by default, we return 2,000 features per dataset. These will be used in downstream analysis, like PCA.
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(pbmc), 10)
# plot variable features with and without labels
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2
Finding variable features for layer counts
When using repel, set xnudge and ynudge to 0 for optimal results
Warning message:
"[1m[22mTransformation introduced infinite values in continuous x-axis"
Warning message:
"[1m[22mTransformation introduced infinite values in continuous x-axis"
Scaling the data
Next, we apply a linear transformation (‘scaling’) that is a standard pre-processing step prior to dimensional reduction techniques like PCA. The ScaleData() function:
- Shifts the expression of each gene, so that the mean expression across cells is 0
- Scales the expression of each gene, so that the variance across cells is 1
- This step gives equal weight in downstream analyses, so that highly-expressed genes do not dominate
- The results of this are stored in
pbmc[["RNA"]]$scale.data - By default, only variable features are scaled.
- You can specify the features argument to scale additional features
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
Centering and scaling data matrix
How can I remove unwanted sources of variation
In Seurat, we also use the ScaleData() function to remove unwanted sources of variation from a single-cell dataset. For example, we could ‘regress out’ heterogeneity associated with (for example) cell cycle stage, or mitochondrial contamination i.e.:
pbmc <- ScaleData(pbmc, vars.to.regress = "percent.mt")
However, particularly for advanced users who would like to use this functionality, we strongly recommend the use of our new normalization workflow, SCTransform(). The method is described in our paper, with a separate vignette using Seurat here. As with ScaleData(), the function SCTransform() also includes a vars.to.regress parameter.
Dimentional Reduction (linear - PCA)
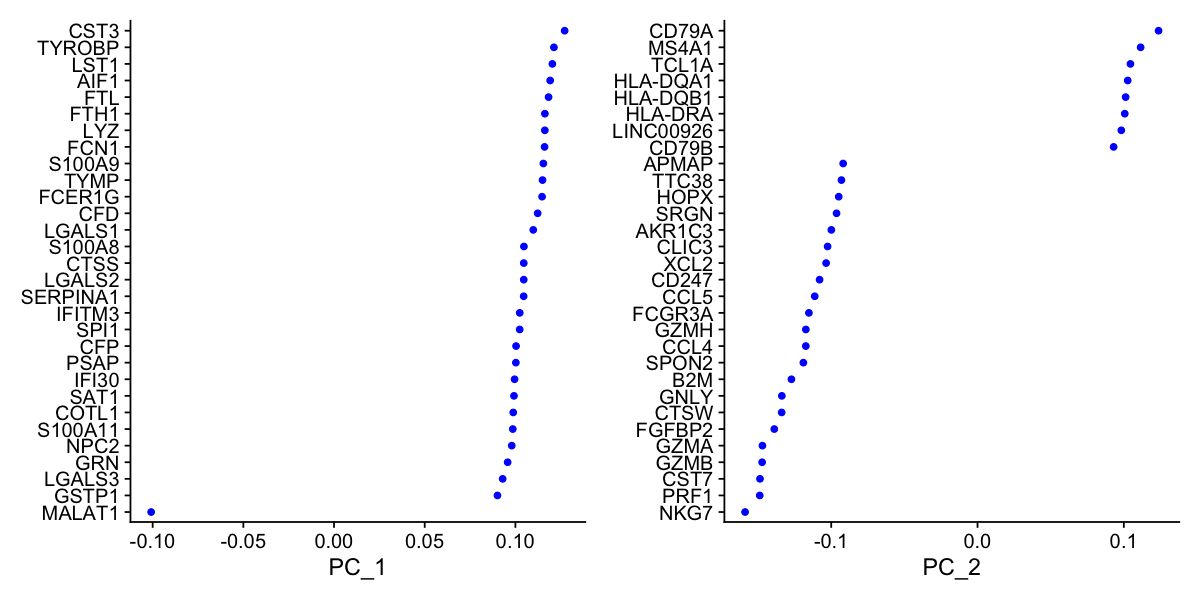
For the first principal components, Seurat outputs a list of genes with the most positive and negative loadings, representing modules of genes that exhibit either correlation (or anti-correlation) across single-cells in the dataset.
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))
PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL, FTH1, LYZ, FCN1, S100A9, TYMP
FCER1G, CFD, LGALS1, S100A8, CTSS, LGALS2, SERPINA1, IFITM3, SPI1, CFP
PSAP, IFI30, SAT1, COTL1, S100A11, NPC2, GRN, LGALS3, GSTP1, PYCARD
Negative: MALAT1, LTB, IL32, IL7R, CD2, B2M, ACAP1, CD27, STK17A, CTSW
CD247, GIMAP5, AQP3, CCL5, SELL, TRAF3IP3, GZMA, MAL, CST7, ITM2A
MYC, GIMAP7, HOPX, BEX2, LDLRAP1, GZMK, ETS1, ZAP70, TNFAIP8, RIC3
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1, HLA-DRA, LINC00926, CD79B, HLA-DRB1, CD74
HLA-DMA, HLA-DPB1, HLA-DQA2, CD37, HLA-DRB5, HLA-DMB, HLA-DPA1, FCRLA, HVCN1, LTB
BLNK, P2RX5, IGLL5, IRF8, SWAP70, ARHGAP24, FCGR2B, SMIM14, PPP1R14A, C16orf74
Negative: NKG7, PRF1, CST7, GZMB, GZMA, FGFBP2, CTSW, GNLY, B2M, SPON2
CCL4, GZMH, FCGR3A, CCL5, CD247, XCL2, CLIC3, AKR1C3, SRGN, HOPX
TTC38, APMAP, CTSC, S100A4, IGFBP7, ANXA1, ID2, IL32, XCL1, RHOC
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1, HLA-DPA1, CD74, MS4A1, HLA-DRB1, HLA-DRA
HLA-DRB5, HLA-DQA2, TCL1A, LINC00926, HLA-DMB, HLA-DMA, CD37, HVCN1, FCRLA, IRF8
PLAC8, BLNK, MALAT1, SMIM14, PLD4, P2RX5, IGLL5, LAT2, SWAP70, FCGR2B
Negative: PPBP, PF4, SDPR, SPARC, GNG11, NRGN, GP9, RGS18, TUBB1, CLU
HIST1H2AC, AP001189.4, ITGA2B, CD9, TMEM40, PTCRA, CA2, ACRBP, MMD, TREML1
NGFRAP1, F13A1, SEPT5, RUFY1, TSC22D1, MPP1, CMTM5, RP11-367G6.3, MYL9, GP1BA
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1, CD74, HIST1H2AC, HLA-DPB1, PF4, SDPR
TCL1A, HLA-DRB1, HLA-DPA1, HLA-DQA2, PPBP, HLA-DRA, LINC00926, GNG11, SPARC, HLA-DRB5
GP9, AP001189.4, CA2, PTCRA, CD9, NRGN, RGS18, CLU, TUBB1, GZMB
Negative: VIM, IL7R, S100A6, IL32, S100A8, S100A4, GIMAP7, S100A10, S100A9, MAL
AQP3, CD2, CD14, FYB, LGALS2, GIMAP4, ANXA1, CD27, FCN1, RBP7
LYZ, S100A11, GIMAP5, MS4A6A, S100A12, FOLR3, TRABD2A, AIF1, IL8, IFI6
PC_ 5
Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY, CCL4, CST7, PRF1, GZMA, SPON2
GZMH, S100A9, LGALS2, CCL3, CTSW, XCL2, CD14, CLIC3, S100A12, RBP7
CCL5, MS4A6A, GSTP1, FOLR3, IGFBP7, TYROBP, TTC38, AKR1C3, XCL1, HOPX
Negative: LTB, IL7R, CKB, VIM, MS4A7, AQP3, CYTIP, RP11-290F20.3, SIGLEC10, HMOX1
LILRB2, PTGES3, MAL, CD27, HN1, CD2, GDI2, CORO1B, ANXA5, TUBA1B
FAM110A, ATP1A1, TRADD, PPA1, CCDC109B, ABRACL, CTD-2006K23.1, WARS, VMO1, FYB
Seurat provides several useful ways of visualizing both cells and features that define the PCA, including VizDimReduction(), DimPlot(), and DimHeatmap()
# Examine and visualize PCA results a few different ways
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
PC_ 1
Positive: CST3, TYROBP, LST1, AIF1, FTL
Negative: MALAT1, LTB, IL32, IL7R, CD2
PC_ 2
Positive: CD79A, MS4A1, TCL1A, HLA-DQA1, HLA-DQB1
Negative: NKG7, PRF1, CST7, GZMB, GZMA
PC_ 3
Positive: HLA-DQA1, CD79A, CD79B, HLA-DQB1, HLA-DPB1
Negative: PPBP, PF4, SDPR, SPARC, GNG11
PC_ 4
Positive: HLA-DQA1, CD79B, CD79A, MS4A1, HLA-DQB1
Negative: VIM, IL7R, S100A6, IL32, S100A8
PC_ 5
Positive: GZMB, NKG7, S100A8, FGFBP2, GNLY
Negative: LTB, IL7R, CKB, VIM, MS4A7
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
DimPlot(pbmc, reduction = "pca") + NoLegend()
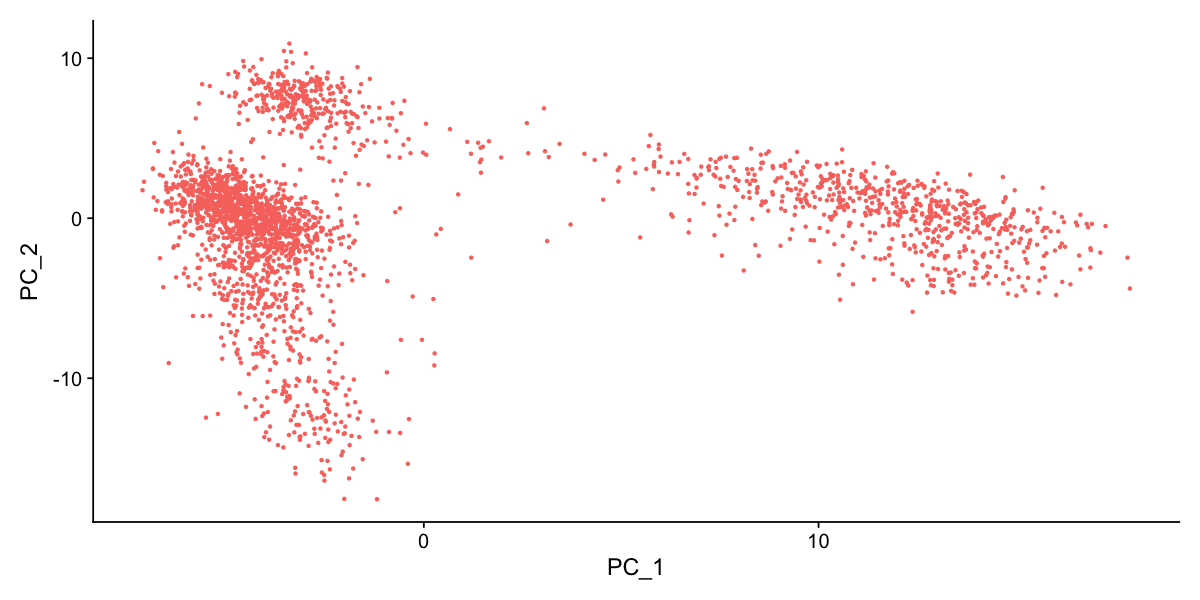
DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)

DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
options(repr.plot.width = 30, repr.plot.height = 50)

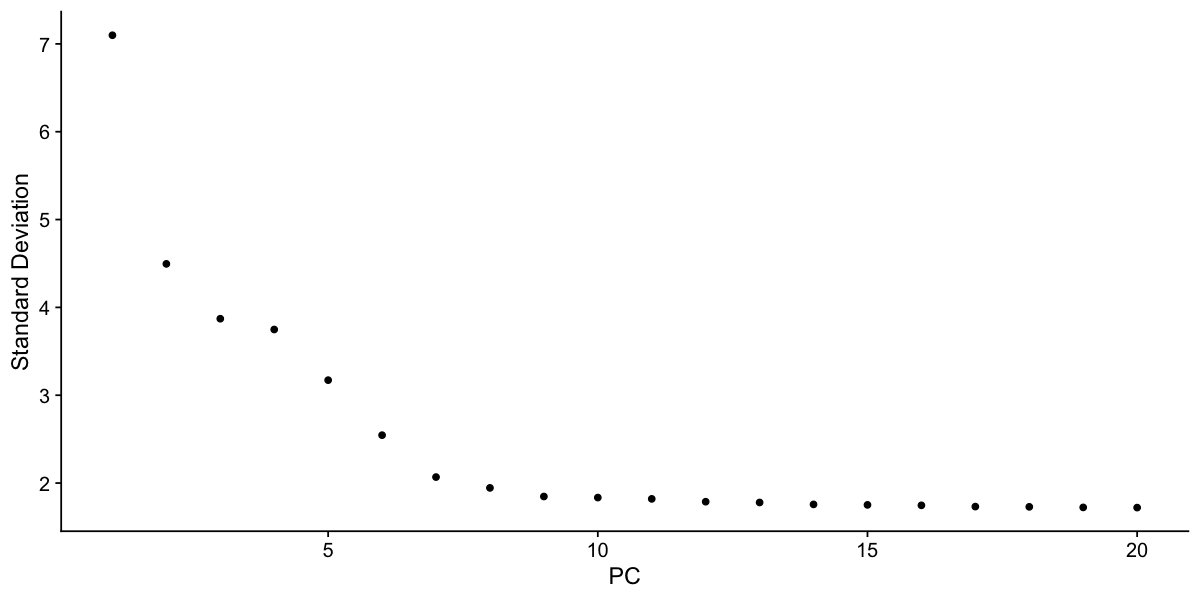
Determine the ‘dimensionality’ of the dataset
Identifying the true dimensionality of a dataset – can be challenging/uncertain for the user. We therefore suggest these multiple approaches for users. The first is more supervised, exploring PCs to determine relevant sources of heterogeneity, and could be used in conjunction with GSEA for example. The second (ElbowPlot) The third is a heuristic that is commonly used, and can be calculated instantly. In this example, we might have been justified in choosing anything between PC 7-12 as a cutoff.
We chose 10 here, but encourage users to consider the following:
- Dendritic cell and NK aficionados may recognize that genes strongly associated with PCs 12 and 13 define rare immune subsets (i.e. MZB1 is a marker for plasmacytoid DCs). However, these groups are so rare, they are difficult to distinguish from background noise for a dataset of this size without prior knowledge.
- We encourage users to repeat downstream analyses with a different number of PCs (10, 15, or even 50!). As you will observe, the results often do not differ dramatically.
- We advise users to err on the higher side when choosing this parameter. For example, performing downstream analyses with only 5 PCs does significantly and adversely affect results.
options(repr.plot.width = 10, repr.plot.height = 5)
ElbowPlot(pbmc)
Cluster the Cells
graph-based clustering approaches
FindClusters(data, resolution = ), good resolution: 0.4-1.2Idents(data)
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
Computing nearest neighbor graph
Computing SNN
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 2638
Number of edges: 95927
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8728
Number of communities: 9
Elapsed time: 0 seconds
# Look at cluster IDs of the first 5 cells
head(Idents(pbmc), 5)
<dl class=dl-inline><dt>AAACATACAACCAC-1</dt><dd>2</dd><dt>AAACATTGAGCTAC-1</dt><dd>3</dd><dt>AAACATTGATCAGC-1</dt><dd>2</dd><dt>AAACCGTGCTTCCG-1</dt><dd>1</dd><dt>AAACCGTGTATGCG-1</dt><dd>6</dd></dl>
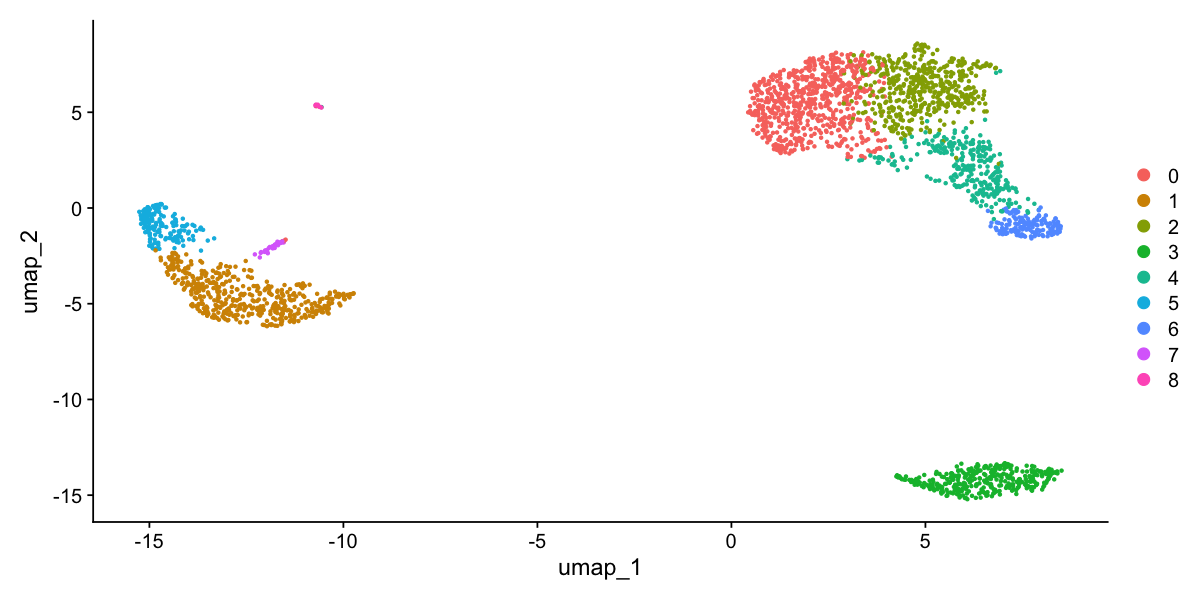
Non-linear dimensional reduction (UMAP/tSNE)
The goal of UMAP and tSNE algorithms is to learn underlying structure in the dataset, in order to place similar cells together in low-dimensional space. Therefore, cells that are grouped together within graph-based clusters determined above should co-localize on these dimension reduction plots.
All visualization techniques have limitations, and cannot fully represent the complexity of the underlying data. In particular, these methods aim to preserve local distances in the dataset (i.e. ensuring that cells with very similar gene expression profiles co-localize), but often do not preserve more global relationships. We encourage users to leverage techniques like UMAP for visualization, but to avoid drawing biological conclusions solely on the basis of visualization techniques.
pbmc <- RunUMAP(pbmc, dims = 1:10)
17:27:29 UMAP embedding parameters a = 0.9922 b = 1.112
17:27:29 Read 2638 rows and found 10 numeric columns
17:27:29 Using Annoy for neighbor search, n_neighbors = 30
17:27:29 Building Annoy index with metric = cosine, n_trees = 50
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
|
17:27:29 Writing NN index file to temp file /var/folders/3j/v5tl8v414pbgrdjndyxmmyx0f7cq_d/T//RtmpSlRmsL/file28f0afaae4
17:27:29 Searching Annoy index using 1 thread, search_k = 3000
17:27:30 Annoy recall = 100%
17:27:30 Commencing smooth kNN distance calibration using 1 thread
with target n_neighbors = 30
17:27:30 Initializing from normalized Laplacian + noise (using RSpectra)
17:27:30 Commencing optimization for 500 epochs, with 105140 positive edges
17:27:32 Optimization finished
# note that you can set `label = TRUE` or use the LabelClusters function to help label
# individual clusters
DimPlot(pbmc, reduction = "umap")
Finding differentially expressed features (cluster biomarkers)
Seurat can help you find markers that define clusters via differential expression (DE). By default, it identifies positive and negative markers of a single cluster (specified in ident.1), compared to all other cells. FindAllMarkers() automates this process for all clusters, but you can also test groups of clusters vs. each other, or against all cells.
# find all markers of cluster 2
cluster2.markers <- FindMarkers(pbmc, ident.1 = 2)
head(cluster2.markers, n = 5)
| p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | |
|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | |
| IL32 | 2.892340e-90 | 1.3070772 | 0.947 | 0.465 | 3.966555e-86 |
| LTB | 1.060121e-86 | 1.3312674 | 0.981 | 0.643 | 1.453850e-82 |
| CD3D | 8.794641e-71 | 1.0597620 | 0.922 | 0.432 | 1.206097e-66 |
| IL7R | 3.516098e-68 | 1.4377848 | 0.750 | 0.326 | 4.821977e-64 |
| LDHB | 1.642480e-67 | 0.9911924 | 0.954 | 0.614 | 2.252497e-63 |
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3))
head(cluster5.markers, n = 5)
| p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | |
|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | |
| FCGR3A | 8.246578e-205 | 6.794969 | 0.975 | 0.040 | 1.130936e-200 |
| IFITM3 | 1.677613e-195 | 6.192558 | 0.975 | 0.049 | 2.300678e-191 |
| CFD | 2.401156e-193 | 6.015172 | 0.938 | 0.038 | 3.292945e-189 |
| CD68 | 2.900384e-191 | 5.530330 | 0.926 | 0.035 | 3.977587e-187 |
| RP11-290F20.3 | 2.513244e-186 | 6.297999 | 0.840 | 0.017 | 3.446663e-182 |
# find markers for every cluster compared to all remaining cells, report only the positive
# ones
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE)
pbmc.markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1)
Calculating cluster 0
Calculating cluster 1
Calculating cluster 2
Calculating cluster 3
Calculating cluster 4
Calculating cluster 5
Calculating cluster 6
Calculating cluster 7
Calculating cluster 8
| p_val | avg_log2FC | pct.1 | pct.2 | p_val_adj | cluster | gene |
|---|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <fct> | <chr> |
| 3.746131e-112 | 1.206019 | 0.912 | 0.592 | 5.137444e-108 | 0 | LDHB |
| 9.571984e-88 | 2.397366 | 0.447 | 0.108 | 1.312702e-83 | 0 | CCR7 |
| 1.154695e-76 | 1.064113 | 0.845 | 0.406 | 1.583548e-72 | 0 | CD3D |
| 1.122405e-54 | 1.043529 | 0.731 | 0.400 | 1.539267e-50 | 0 | CD3E |
| 1.354319e-51 | 2.136530 | 0.342 | 0.103 | 1.857312e-47 | 0 | LEF1 |
| 1.942957e-47 | 1.198913 | 0.629 | 0.359 | 2.664571e-43 | 0 | NOSIP |
| 2.806087e-44 | 1.526200 | 0.443 | 0.185 | 3.848268e-40 | 0 | PIK3IP1 |
| 6.269443e-43 | 1.985307 | 0.330 | 0.112 | 8.597914e-39 | 0 | PRKCQ-AS1 |
| 1.161169e-40 | 2.696721 | 0.200 | 0.040 | 1.592427e-36 | 0 | FHIT |
| 1.339878e-34 | 1.956368 | 0.268 | 0.087 | 1.837508e-30 | 0 | MAL |
| 1.995541e-34 | 1.337712 | 0.393 | 0.177 | 2.736686e-30 | 0 | TCF7 |
| 3.577936e-33 | 2.819950 | 0.155 | 0.029 | 4.906781e-29 | 0 | LINC00176 |
| 1.577717e-30 | 2.212287 | 0.158 | 0.033 | 2.163682e-26 | 0 | NELL2 |
| 6.262951e-30 | 2.364034 | 0.247 | 0.085 | 8.589011e-26 | 0 | LDLRAP1 |
| 1.463401e-27 | 1.639526 | 0.199 | 0.060 | 2.006908e-23 | 0 | TRABD2A |
| 5.212157e-26 | 1.101997 | 0.404 | 0.209 | 7.147951e-22 | 0 | LEPROTL1 |
| 4.951637e-24 | 3.017245 | 0.102 | 0.016 | 6.790676e-20 | 0 | ADTRP |
| 3.299109e-23 | 1.713325 | 0.219 | 0.081 | 4.524398e-19 | 0 | OXNAD1 |
| 1.332403e-21 | 1.053905 | 0.360 | 0.185 | 1.827258e-17 | 0 | RGCC |
| 4.865492e-21 | 2.238698 | 0.123 | 0.030 | 6.672535e-17 | 0 | EPHX2 |
| 3.332800e-20 | 2.528041 | 0.118 | 0.029 | 4.570602e-16 | 0 | SCGB3A1 |
| 5.243698e-20 | 1.616091 | 0.189 | 0.069 | 7.191208e-16 | 0 | SH3YL1 |
| 1.825309e-19 | 1.057635 | 0.444 | 0.283 | 2.503228e-15 | 0 | NDFIP1 |
| 2.039629e-19 | 1.108486 | 0.425 | 0.258 | 2.797147e-15 | 0 | C12orf57 |
| 2.419804e-19 | 2.191440 | 0.120 | 0.031 | 3.318519e-15 | 0 | C14orf64 |
| 4.620287e-19 | 2.163959 | 0.124 | 0.034 | 6.336262e-15 | 0 | RP11-664D1.1 |
| 5.361879e-19 | 1.358684 | 0.276 | 0.137 | 7.353281e-15 | 0 | FLT3LG |
| 7.582402e-19 | 1.092250 | 0.174 | 0.062 | 1.039851e-14 | 0 | NGFRAP1 |
| 1.480780e-18 | 1.046770 | 0.370 | 0.208 | 2.030742e-14 | 0 | RHOH |
| 2.799851e-17 | 2.197118 | 0.114 | 0.031 | 3.839715e-13 | 0 | APBA2 |
| ... | ... | ... | ... | ... | ... | ... |
| 0.006937108 | 2.253826 | 0.077 | 0.008 | 1 | 8 | TRIP6 |
| 0.006937108 | 2.126115 | 0.077 | 0.008 | 1 | 8 | MCU |
| 0.006937108 | 2.304901 | 0.077 | 0.008 | 1 | 8 | MSANTD4 |
| 0.007049947 | 4.060892 | 0.231 | 0.062 | 1 | 8 | DAPP1 |
| 0.007410831 | 2.000718 | 0.154 | 0.028 | 1 | 8 | PCIF1 |
| 0.007452686 | 1.894379 | 0.615 | 0.431 | 1 | 8 | TUBA1B |
| 0.007472699 | 5.008332 | 0.077 | 0.008 | 1 | 8 | STK3 |
| 0.007523382 | 4.211537 | 0.077 | 0.008 | 1 | 8 | TSPAN15 |
| 0.007526733 | 2.943000 | 0.154 | 0.029 | 1 | 8 | CYB5D2 |
| 0.007677286 | 3.818398 | 0.077 | 0.008 | 1 | 8 | DAG1 |
| 0.007758834 | 1.000930 | 0.769 | 0.753 | 1 | 8 | TSC22D3 |
| 0.007843344 | 2.093528 | 0.154 | 0.028 | 1 | 8 | ABHD11 |
| 0.008041631 | 3.711957 | 0.154 | 0.029 | 1 | 8 | SVIL |
| 0.008207813 | 3.377103 | 0.231 | 0.063 | 1 | 8 | MIR4435-1HG |
| 0.008506282 | 1.952535 | 0.154 | 0.029 | 1 | 8 | TBC1D9B |
| 0.008511611 | 1.017637 | 0.846 | 0.782 | 1 | 8 | SRP14 |
| 0.008603911 | 2.644436 | 0.077 | 0.008 | 1 | 8 | CTNS |
| 0.008661409 | 2.187964 | 0.077 | 0.008 | 1 | 8 | XRCC4 |
| 0.008661409 | 2.099569 | 0.077 | 0.008 | 1 | 8 | C16orf86 |
| 0.008661409 | 2.302028 | 0.077 | 0.008 | 1 | 8 | VPS9D1 |
| 0.008661409 | 2.129861 | 0.077 | 0.008 | 1 | 8 | KLF16 |
| 0.008869496 | 2.511113 | 0.692 | 0.449 | 1 | 8 | TRAPPC1 |
| 0.009164182 | 5.146753 | 0.077 | 0.009 | 1 | 8 | GSTM4 |
| 0.009164182 | 5.017036 | 0.077 | 0.009 | 1 | 8 | MRPS24 |
| 0.009164182 | 5.111850 | 0.077 | 0.009 | 1 | 8 | PTPRJ |
| 0.009164182 | 4.822627 | 0.077 | 0.009 | 1 | 8 | CDK2 |
| 0.009201825 | 2.670857 | 0.154 | 0.030 | 1 | 8 | BCL2L1 |
| 0.009334658 | 2.912823 | 0.154 | 0.030 | 1 | 8 | ETV6 |
| 0.009838577 | 3.310543 | 0.077 | 0.009 | 1 | 8 | VWA8 |
| 0.009946527 | 2.067077 | 0.538 | 0.309 | 1 | 8 | AP2M1 |
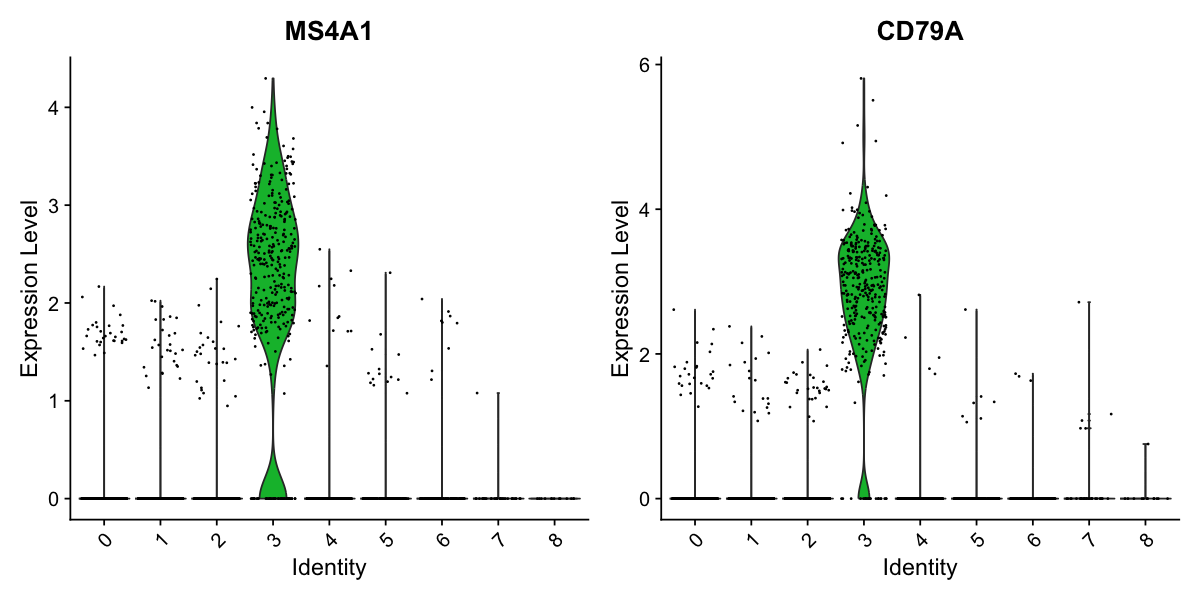
cluster0.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
# you can plot raw counts as well
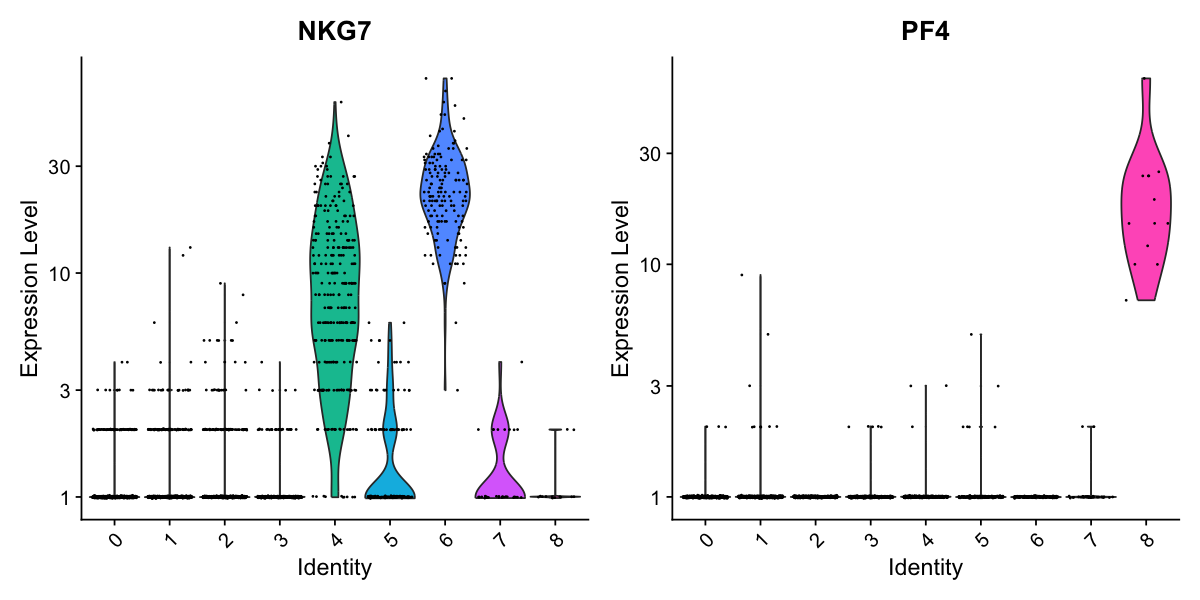
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)
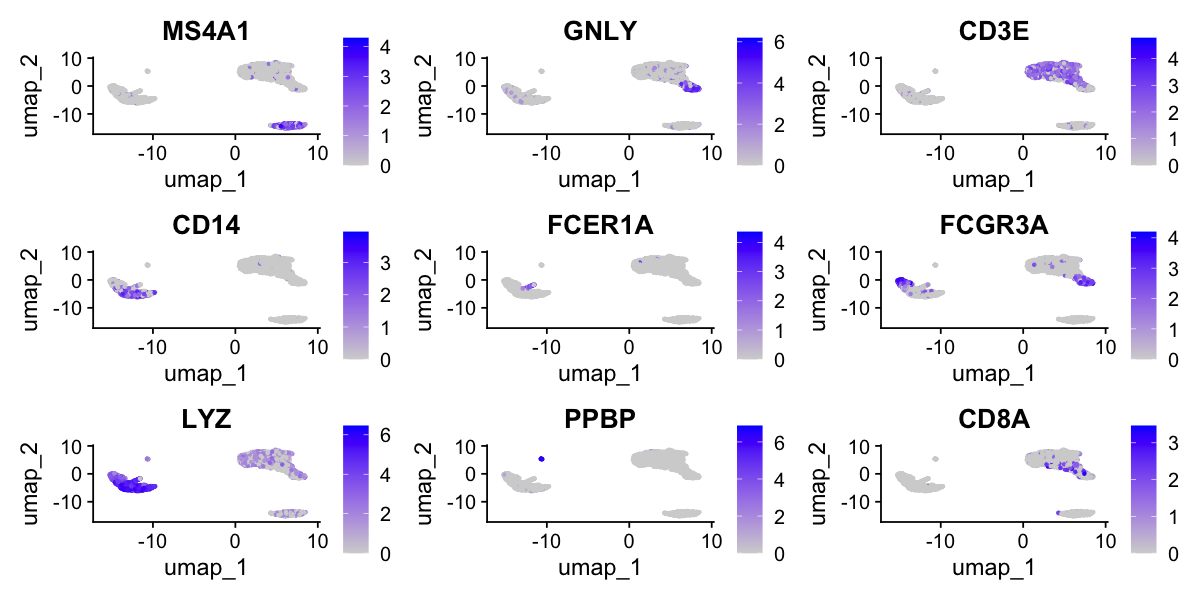
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
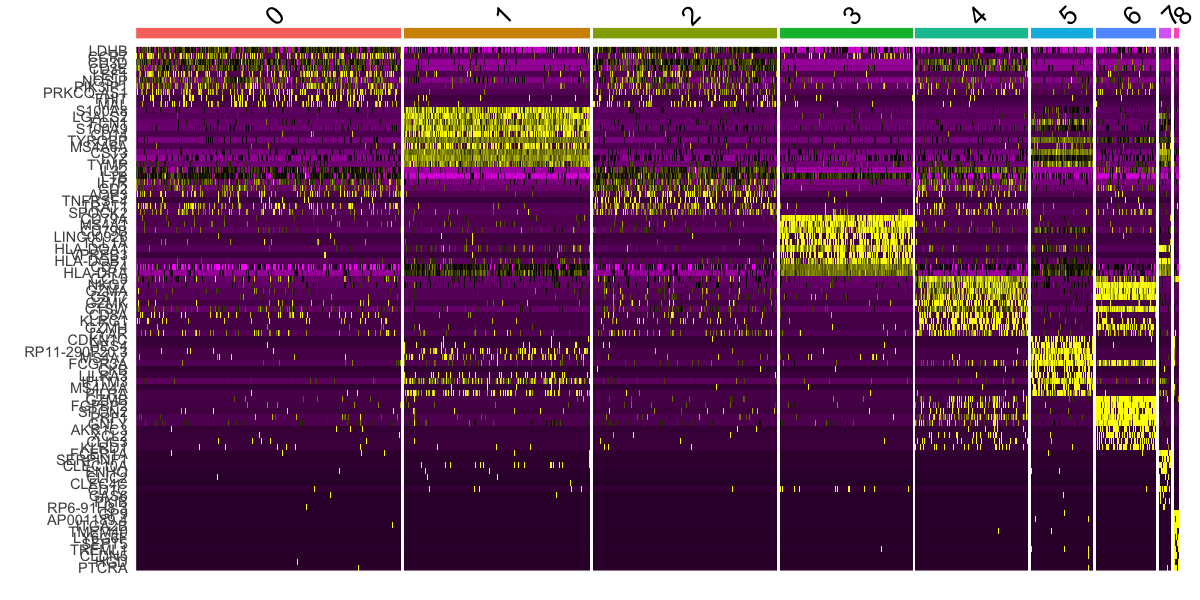
DoHeatmap() generates an expression heatmap for given cells and features. In this case, we are plotting the top 20 markers (or all markers if less than 20) for each cluster.
pbmc.markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 10) %>%
ungroup() -> top10
DoHeatmap(pbmc, features = top10$gene) + NoLegend()
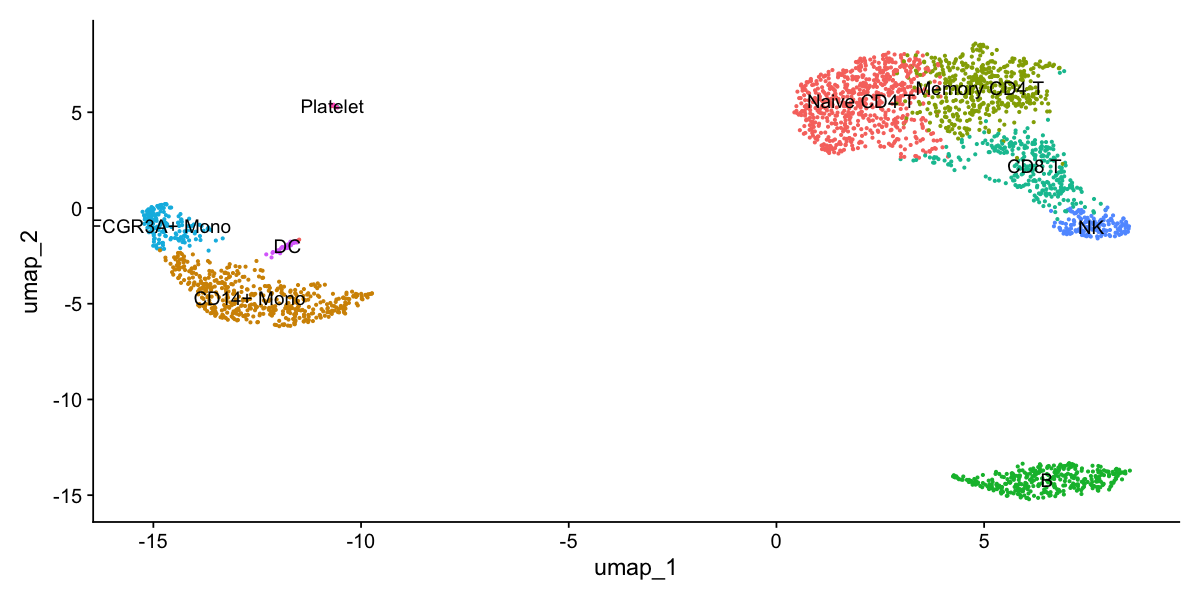
Assigning cell type identity to clusters
Fortunately in the case of this dataset, we can use canonical markers to easily match the unbiased clustering to known cell types:
Cluster ID Markers Cell Type 0 IL7R, CCR7 Naive CD4+ T 1 CD14, LYZ CD14+ Mono 2 IL7R, S100A4 Memory CD4+ 3 MS4A1 B 4 CD8A CD8+ T 5 FCGR3A, MS4A7 FCGR3A+ Mono 6 GNLY, NKG7 NK 7 FCER1A, CST3 DC 8 PPBP Platelet
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
library(ggplot2)
plot <- DimPlot(pbmc, reduction = "umap", label = TRUE, label.size = 4.5) + xlab("UMAP 1") + ylab("UMAP 2") +
theme(axis.title = element_text(size = 18), legend.text = element_text(size = 18)) + guides(colour = guide_legend(override.aes = list(size = 10)))
ggsave(filename = "../output/images/pbmc3k_umap.jpg", height = 7, width = 12, plot = plot, quality = 50)
saveRDS(pbmc, file = "../output/pbmc3k_final.rds")
sessionInfo()
R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Sonoma 14.3.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] C
time zone: Australia/Brisbane
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggplot2_3.4.4 gridExtra_2.3 patchwork_1.2.0 Seurat_5.0.1
[5] SeuratObject_5.0.1 sp_2.1-3 dplyr_1.1.4
loaded via a namespace (and not attached):
[1] matrixStats_1.2.0 spatstat.sparse_3.0-3 httr_1.4.7
[4] RColorBrewer_1.1-3 repr_1.1.6 tools_4.3.2
[7] sctransform_0.4.1 utf8_1.2.4 R6_2.5.1
[10] lazyeval_0.2.2 uwot_0.1.16 withr_3.0.0
[13] progressr_0.14.0 textshaping_0.3.7 cli_3.6.2
[16] spatstat.explore_3.2-6 fastDummies_1.7.3 labeling_0.4.3
[19] spatstat.data_3.0-4 ggridges_0.5.6 pbapply_1.7-2
[22] systemfonts_1.0.5 pbdZMQ_0.3-11 R.utils_2.12.3
[25] parallelly_1.37.0 generics_0.1.3 ica_1.0-3
[28] spatstat.random_3.2-2 Matrix_1.6-5 fansi_1.0.6
[31] abind_1.4-5 R.methodsS3_1.8.2 lifecycle_1.0.4
[34] Rtsne_0.17 grid_4.3.2 promises_1.2.1
[37] crayon_1.5.2 miniUI_0.1.1.1 lattice_0.21-9
[40] cowplot_1.1.3 pillar_1.9.0 future.apply_1.11.1
[43] codetools_0.2-19 leiden_0.4.3.1 glue_1.7.0
[46] getPass_0.2-4 data.table_1.15.0 vctrs_0.6.5
[49] png_0.1-8 spam_2.10-0 gtable_0.3.4
[52] mime_0.12 survival_3.5-7 ellipsis_0.3.2
[55] fitdistrplus_1.1-11 ROCR_1.0-11 nlme_3.1-163
[58] RcppAnnoy_0.0.22 irlba_2.3.5.1 KernSmooth_2.23-22
[61] colorspace_2.1-0 tidyselect_1.2.0 compiler_4.3.2
[64] plotly_4.10.4 scales_1.3.0 lmtest_0.9-40
[67] stringr_1.5.1 digest_0.6.34 goftest_1.2-3
[70] spatstat.utils_3.0-4 htmltools_0.5.7 pkgconfig_2.0.3
[73] base64enc_0.1-3 fastmap_1.1.1 rlang_1.1.3
[76] htmlwidgets_1.6.4 shiny_1.8.0 farver_2.1.1
[79] zoo_1.8-12 jsonlite_1.8.8 R.oo_1.26.0
[82] magrittr_2.0.3 dotCall64_1.1-1 IRkernel_1.3.2
[85] munsell_0.5.0 Rcpp_1.0.12 reticulate_1.35.0
[88] stringi_1.8.3 MASS_7.3-60 plyr_1.8.9
[91] parallel_4.3.2 listenv_0.9.1 ggrepel_0.9.5
[94] deldir_2.0-2 IRdisplay_1.1 splines_4.3.2
[97] tensor_1.5 igraph_2.0.2 uuid_1.2-0
[100] spatstat.geom_3.2-8 RcppHNSW_0.6.0 reshape2_1.4.4
[103] evaluate_0.23 httpuv_1.6.14 RANN_2.6.1
[106] tidyr_1.3.1 purrr_1.0.2 polyclip_1.10-6
[109] future_1.33.1 scattermore_1.2 xtable_1.8-4
[112] RSpectra_0.16-1 later_1.3.2 ragg_1.2.7
[115] viridisLite_0.4.2 tibble_3.2.1 cluster_2.1.4
[118] globals_0.16.2